![]()
Research Interests
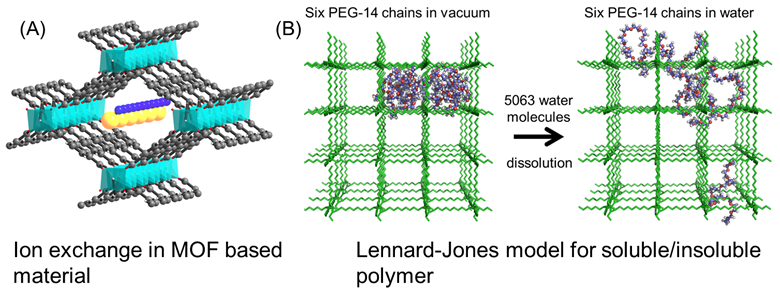
Canonical molecular dynamics simulations are performed to
investigate behavior of single-chain and multiple-chain polyethylene glycol
(PEG) contained within a cubic framework spanned by polyethylene (PE)
Lennard-Jones chains. This simple model is first of its kind to study the
chemical physics of polymer threaded organic frameworks which are materials with
potential applications in catalysis and separation processes. Novel ion-exchange
polyelectrolyte~metal organic framework (MOF) materials achieve both superior
ion exchange kinetics and high ion selectivity. Direct detection of single ion
motion in solution is beyond the ability of experimental techniques. Molecular
dynamics simulations combined with umbrella sampling are used to explain the
cation/anion exchange process occurred within polyelectrolyte~MOF. We use
molecular dynamics simulations to screen the matching polyelectrolyte and MOF
composites and to assist the following experimental material design.